
Quelle place pour les études de qualité de vie aux côtés de l’ « essai contrôlé randomisé » ?

L’ « essai contrôlé randomisé », reine des preuves…
L’« essai contrôlé randomisé » (ECR) est le meilleur moyen d’évaluer les effets des traitements thérapeutiques comparés les uns aux autres.
L’ECR constitue l’une des phases de la recherche dite « interventionnelle » qui impliquent une intervention non dénuée de risque pour les personnes qui y participent, et non justifiée par leur prise en charge habituelle.
Une étude ou essai contrôlé randomisé est un protocole expérimental ayant pour but d’évaluer l’efficacité d’une thérapie, d’une action de prévention ou d’un médicament. Elle compare un groupe expérimental dit groupe d’intervention à qui l’on administre le traitement standard ou prenant un placebo. Un critère de jugement est fixé en amont ; il peut être objectif (taux de cholestérol, régression des symptômes…) ou subjectif (mesure de l’anxiété, de la douleur, qualité de vie ressentie…).
Deux critères essentiels doivent être respectés pour prétendre à un niveau de preuve suffisant pour confirmer l’efficacité d’un traitement :
- L’attribution dans un groupe se fait de façon aléatoire par tirage au sort (randomisation).
- La répartition des groupes se fait à l’insu des participants (étude en aveugle) et éventuellement des cliniciens (double aveugle).
Une étude randomisée permet ainsi d’établir formellement un lien de causalité, sous réserve qu’aucune source de biais secondaire n’ait été introduite en cours d’essai (comme par exemple des procédures de suivi différentes dans les deux groupes). L’étude randomisée correspond à la phase 3 des essais cliniques nécessaires à la mise sur le marché d’un nouveau médicament.
- La phase 1 teste la tolérance chez le sujet sain,
- la phase 2 évalue l’efficacité sur des sujets malades,
- l’essai randomisé dit de phase 3 permet d’amener la preuve de l’intérêt ou de la supériorité du nouveau traitement par rapport à un équivalent ou un placebo,
- la phase 4 est destinée à la pharmacovigilance et à la détection d’éventuels effets secondaires.
Les recherches interventionnelles doivent recevoir l’avis favorable d’un comité de protection des personnes (CPP) avant de débuter.
…qui a pourtant lui-même ses limites
Plusieurs facteurs peuvent limiter la validité des ECR, notamment :
- Ils peuvent inclure des patients dont le pronostic est meilleur que la moyenne.
- Ils peuvent exclure des femmes, des enfants, des personnes âgées ou des patients souffrant de plusieurs pathologies.
- Ils ne reflètent pas les conditions réelles d’utilisation des produits par les patients.
- Ils ne sont pas toujours possibles lorsque la pathologie étudiée est rare.
- Ils ne permettent pas d’évaluer l’ensemble des effets indésirables qui peuvent intervenir avec plusieurs années de recul.
Les études randomisées sont très coûteuses et difficiles à conduire à grande échelle.
Les études observationnelles, complémentaires des ECR
Dans les faits, la plupart des études sur les produits de santé sont des études non expérimentales, dites « observationnelles ».
Les études observationnelles peuvent être descriptives ou analytiques et procèdent de la recherche dite « non interventionnelle » : elles ne modifient pas la prise en charge des participants et tous les actes pratiqués et tous les produits utilisés le sont dans le cadre d’une prise en charge habituelle.
Une étude observationnelle tire des conclusions sur l’effet possible d’un traitement sur les participants, lorsque l’affectation des participants à un groupe de traitement par rapport à un groupe de contrôle n’est pas du ressort de l’investigateur.
- Lorsque la mise en place d’un groupe traitement et d’un groupe contrôle n’est pas possible, une étude observationnelle permet néanmoins d’obtenir des informations intéressantes sur les effets d’un traitement.
Dans certains cas, les études observationnelles constituent la méthodologie la plus appropriée, si la pathologie étudiée est rare, par exemple.
- Les études non interventionnelles permettent par ailleurs d’observer des effets impossibles à évaluer par l’essai contrôlé randomisé (ECR) pour des raisons éthiques. Par exemple, si l’effet d’un facteur de risque environnemental tel que l’amiante est étudié, il ne serait pas éthique d’exposer délibérément les participants à l’amiante.
- La recherche observationnelle permet d’évaluer des améliorations de l’état de santé, de la survie et de la qualité de vie des personnes ayant suivi une thérapie ou une action de prévention. Elle peut aussi observer des bénéfices complémentaires auprès des familles et des proches, voire des économies.
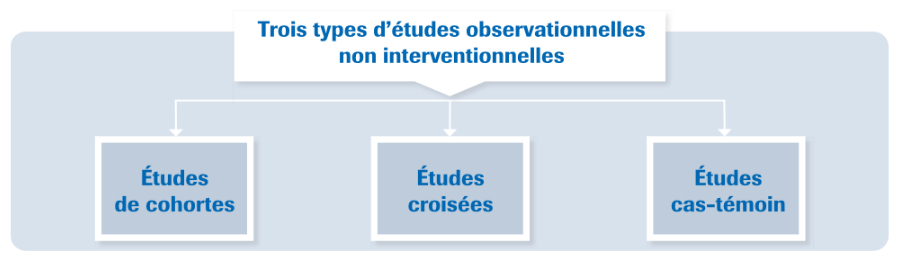
Les recherches non interventionnelles basées sur l’observation (analyses rétrospectives de données collectées à partir de la pratique clinique quotidienne, comparaison d’efficacité de pratiques de soins différentes entre établissements de santé, etc.) peuvent être mises en place sans autorisation éthique préalable, ni du CPP, ni de l’ANSM.
L’ANSM demande peu d’études observationnelles aux industriels, elle en réalise directement ou en fait réaliser par des équipes de recherche.
La HAS commande les études post AMM (Autorisation de Mise sur le Marché dite post-inscription) aux laboratoires et valide les protocoles en amont des études.
Les études de qualité de vie dans tout cela ?
Si l’ECR est réputé être le « gold standard » de la médecine fondée sur les preuves, les enseignements qu’il apporte doivent être complétés par des études en vie réelle qui rendent compte de l’impact du médicament sur la durée et sur la qualité de vie des patients. Il n’y a en effet pas d’opposition entre recherche observationnelle et recherche interventionnelle mais bien une complémentarité entre les deux méthodologies.
Si de nombreux pays se sont d’ores et déjà engagés sur le recueil de données pertinentes pour renforcer la robustesse méthodologique de l’évaluation de l’expérience patient, la France tarde à déployer cette démarche à grande échelle, à diffuser une nouvelle culture de l’évaluation fondée sur des approches renouvelées de l’Evidence Based Medicine dont les études de qualité de vie font partie.
Intimement liée à l’amélioration de la qualité de vie, la qualité des soins devrait imposer de nouveaux arbitrages sur les stratégies thérapeutiques. De nouvelles preuves sont attendues, pour comparer les traitements entre eux du point de vue de leur efficacité et des coûts / bénéfices et pour transformer notre système de santé au service de l’ « humain ».
Dans ce contexte, les patients, eux-mêmes dépositaires de très nombreuses données utiles, deviennent des acteurs de la recherche en santé.
En quoi les data peuvent améliorer les connaissances sur la qualité de vie des patients ?
Dans la lettre de mission adressée le 6 juillet 2016 à Madame Dominique Polton et au professeur Bernard Bégaud, Madame Touraine, alors ministre de la Santé, partait du constat suivant:
Le besoin de données en vie réelle est d’autant plus crucial que nous sommes dans un contexte de dynamique d’innovation soutenue, qui se traduit par des avancées bénéfiques pour les patients, mais coûteuses pour le système de santé. L’effort financier consenti par la collectivité appelle une évaluation rigoureuse des performances attendus du traitement au vu des essais cliniques, mais aussi la confirmation de ces résultats à l’usage dans la pratique courante, car l’expérience montre que l’efficacité en vie réelle peut différer de celle observée dans des conditions expérimentales (conditions d’utilisation et populations différentes, observance moindre…).
Les évolutions actuelles ne font que renforcer cette nécessité d’un suivi attentif des performances du médicament en vie réelle : arrivées précoces de produits après des essais de phase II, AMM conditionnelles et adaptive pathways, faibles niveaux de preuve du fait de petites populations dans les essais (médicaments orphelins, thérapies ciblées…).
Ainsi, dans plusieurs pays, le financement de certains médicaments coûteux est conditionné à la mise en place des registres permettant de documenter leur usage, de produire des connaissances pour lever les incertitudes qui subsistent lors de l’évaluation en primo-inscription, et dans certains cas de conclure avec les industriels des contrats de performance.
Invitée à recenser les outils d’information déjà mis en place par différents acteurs, à identifier les domaines prioritaires en termes de besoin de suivi en vie réelle, à envisager des options pour la surveillance des médicaments post AMM, la mission proposait notamment de garantir la qualité et de renforcer la confiance dans les études observationnelles.
Si, du fait de la relative facilité de mise en œuvre, un nombre non négligeable d’études observationnelles de faible qualité sont publiées ou rendues publiques, il est essentiel de rappeler deux évidences : c’est l’observation, et donc l’approche observationnelle, qui a été depuis toujours à la base des avancées scientifiques les plus importantes, particulièrement dans le domaine de la santé; comme pour toutes les méthodes et sciences d’investigation, la maitrise de l’approche pharmaco-épidémiologique, discipline récente, demande une formation sérieuse et une expérience avérée.
La recevabilité des études observationnelles dépend donc de leur validité.
Selon la mission, plusieurs éléments sont de nature à garantir la qualité des études observationnelles, pour que l’on puisse utiliser leurs résultats avec confiance :
S’il est admis que les données de vie réelle peuvent améliorer les connaissances sur la qualité de vie des patients, elles influenceront l’évaluation des produits de santé si et seulement si ces études observationnelles respectent des référentiels de méthodes qui dissiperont toute défiance vis-à-vis des résultats obtenus.
En savoir plus
Rapport réalisé par Bernard Bégaud, Dominique Polton, Franck von Lennep à la demande de Madame la Ministre de la santé Marisol Touraine
Lire aussi :
Atelier 1
Participer à l’évaluation de l’innovation thérapeutique